近日,伟德国际victor1946教师尹志平联合南丹麦大学吴昌柱课题组在国际化学顶级期刊Angew. Chem.上在线发表题为Photoenzymatic Aliphatic C-Br Activation for Enantioselective Synthesis of γ-Stereogenic Nitriles from Alkenes (doi.org/10.1002/anie.202519301) 的最新研究成果,伟德victor1946为该论文的第一通讯单位。该研究得到江苏省自然科学基金青年项目及丹麦嘉士伯基金会的资助。
酶促直接引入氰基的策略仍不成熟,限制了药物和功能材料中手性腈衍生物的合成。本文发展了一种光酶促γ-氢氰化策略,实现了对这一瓶颈的突破。通过工程化烯还原酶,在可见光激发下,还原型黄素辅因子可高效活化具有挑战性的脂肪族C─Br键(如溴乙腈),生成的氰甲基自由基在酶活性位点内被精确限制并定向,与α-甲基苯乙烯偶联。该体系展现出显著的立体控制能力,活性位点同时负责自由基生成与后续对映选择性的氢原子转移,可生成多种γ-立体中心手性腈,产率最高达93%,对映选择性可达94% ee,而此类反应对传统过渡金属催化具有相当挑战性。该研究不仅提供了一个稳健的非对称氰烷基化平台,也推动了光酶促催化的发展,首次证明非活化烷基溴化物可作为可控成键的有效自由基前体。
光酶促催化通过结合光化学的多样反应性与生物催化的精确选择性,已革新了非对称合成,并在黄素依赖型烯还原酶的基础上实现了对自由基转化的原子级控制。尽管该领域已在活化α-羰基卤代物、实现分子间加成等方面取得进展,并扩展到ThDP、PLP依赖酶及人工光酶体系,但对无活化烷基C–Br键的直接活化仍未实现,而这类底物比碘代物更易获得且结构更丰富。另一方面,氰基因其独特的电子性质和合成可转化性,是70余种临床小分子药物中的关键结构,但现有方法难以在无活化烯烃上直接、对映选择性地构建带γ-立体中心的烷基腈。基于上述挑战,作者提出利用光酶促催化开发可在可见光下活化惰性C–Br键的工程化烯还原酶,同时精确控制自由基的立体化学,实现以溴乙腈为氰化剂,将α-甲基苯乙烯转化为γ-立体中心腈类,从而拓展手性腈的合成工具箱并开启烷基自由基光酶促化学的新方向(方案1)。
研究对十二种烯还原酶的初筛显示其活性与立体选择性差异显著;PETNR野生型因兼具较高转化率与对映选择性而被选为工程化基础。通过分子对接与活性位点分析,研究锁定H182、H185为调控关键,并对其进行系统突变,其中H182M表现最佳,使产率升至93%、ee达94%。光照、酶环境与NADPH均被证明对反应必不可少,蛋白支架对自由基过程尤为关键。在优化条件下,工程化PETNR可高效催化α-甲基苯乙烯与溴乙腈的γ-氰基化。底物拓展表明该体系兼容多种取代烯烃,能耐受供/吸电子基团、游离酚羟基及扩展芳环,普遍获得良好产率和高ee;但邻位位阻、较大烷基取代或非芳香烯烃会显著降低反应效率,强调苯环在该自由基立体控制过程中的关键作用(表1-2)。

方案1光酶促催化及有机腈类化合物的合成

表1 条件优化
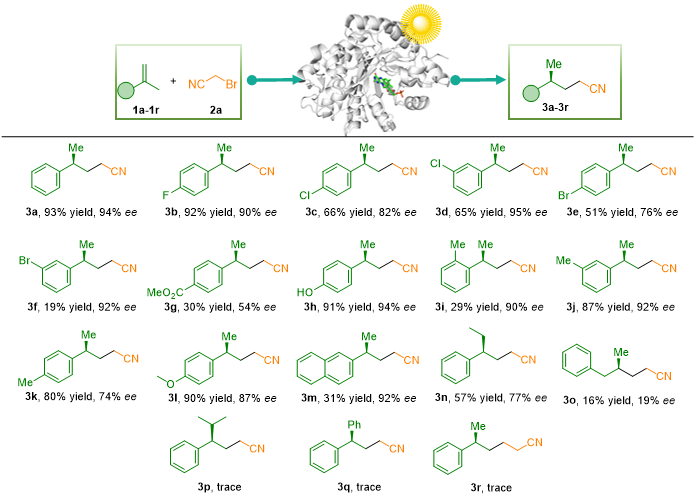
表2光酶促合成有机腈类化合物的底物适用性

方案2催化循环与电荷转移复合物
该研究验证了反应的可放大性,并证明得到的对映富集γ-氰基产物可通过酯化或酰胺化实现多样化转化且保持立体纯度。通过光谱分析、自由基捕获和对照实验,作者揭示了反应机理:还原态FMN在光照下与底物形成电荷转移复合物,产生关键的氰甲基自由基,该自由基加成至烯烃后形成稳定的苄基自由基中间体,随后经FMNsq的立体选择性氢原子转移构建出γ-立体中心,并完成催化循环(方案2)。最终构建的酶促光催化体系能够有效实现惰性C–Br键活化,实现高产率、高对映选择性的γ-氰基化,为构建手性腈类化合物提供了实用且可拓展的策略,并为开发新的C–Br光酶促活化方法奠定基础。


 伟德国际victor1946公众号
伟德国际victor1946公众号
